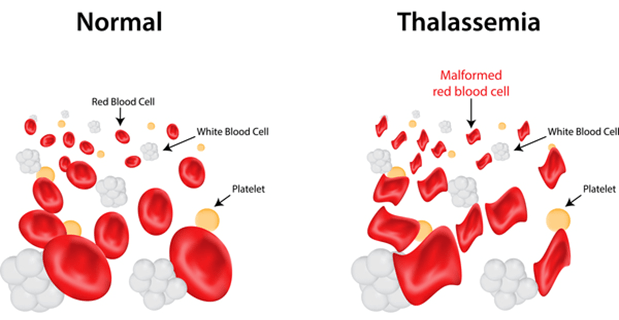
Thalassemia is an inherited blood condition. If you have thalassemia, your body has fewer red blood cells and less hemoglobin than it should. Thalassemia is divided into alpha-thalassemia and beta-thalassemia, which are called for abnormalities that may arise in these protein chains.
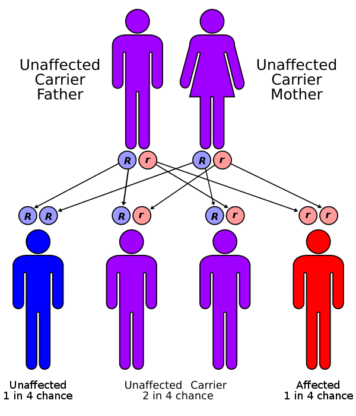
Hemoglobin molecules are made up of alpha and beta chains, which are susceptible to mutations. The production of either the alpha or beta chains is diminished in thalassemia, resulting in alpha- or beta-thalassemia. The severity of alpha-thalassemia is determined by the number of gene mutations you receive from your parents. The more mutated genes you have, the worse your thalassemia will be. The severity of beta-thalassemia is determined by the section of the hemoglobin molecule that is damaged.
Fatigue, anemia, weakness, pale or yellowish complexion, slow development, abdominal enlargement, dark urine, and deformities of the facial bones are all symptoms of thalassemia. A family history of thalassemia is one factor that raises your risk of thalassemia. Thalassemia is more common in Pakistan due to the high ratio of cousin marriages. Thalassemia may be prevented by having a thalassemia blood test done before getting married. It is also important to consider your family background.
You may get testing to see whether your baby will have the issue if you are pregnant or attempting to conceive. Genetic testing may reveal if you or your spouse possesses any of the thalassemia-causing genes. A chorionic villus sample examines a little bit of the placenta to discover whether a newborn has the thalassemia genes. This test is normally performed during the 11th week of pregnancy. Amniocentesis is a procedure that examines the fluid around an unborn fetus. This test is normally performed during the 16th week of pregnancy.

The only currently available permanent solution for thalassemia is a stem cell or bone marrow transplant, although they are not frequently done due to the severe hazards involved. Bone marrow, the spongy substance located in the center of certain bones, produces stem cells, which can grow into many kinds of blood cells. Blood transfusions and iron chelation are standard therapies for people with thalassemia major.
Anemia may be minor or severe in people with thalassemia. Severe anemia and iron overload due to multiple blood transfusions may cause organ damage and even death. Iron overload can damage any organ of the body; most common and life-threatening are liver failure, heart, bony changes, and thyroid gland. In such cases liver and heart cannot perform its functions so the patient may die because of the complications.
| Cookie | Duration | Description |
|---|---|---|
| cookielawinfo-checkbox-analytics | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics". |
| cookielawinfo-checkbox-functional | 11 months | The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional". |
| cookielawinfo-checkbox-necessary | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary". |
| cookielawinfo-checkbox-others | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other. |
| cookielawinfo-checkbox-performance | 11 months | This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance". |
| viewed_cookie_policy | 11 months | The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data. |